Input structure
clTEM supports opening .xyz and .cif files. However, some modifications to the .xyz format are supported so that occupancy and thermal parameters can be included.
.cif
The .cif format is a ubiquitous crystallographic file format that is useful for simulating bulk, periodic materials. However, the format is not very will standardised, and many .cif files will not open in clTEM. clTEM will warn you if it cannot read the .cif input and will try and correct some of the typical mistakes found in .cif files, though there is no guarantee the ‘fixed’ output will be correct. The way clTEM attempts to read .cif files is outlines as:
- The symmetry is determined from the
_symmetry_equiv_pos_as_xyzdata (i.e. not the space group name). - The atom types are determined from the
_atom_site_type_symboldata (commonly only the label will be defined). - Thermal parameters will not be interpreted from B-factors, only defined u parameters (applied to atoms with matching labels).
- Currently, anisotropic u values will be applied along the simulation’s cartesian axes.
.cif dialog
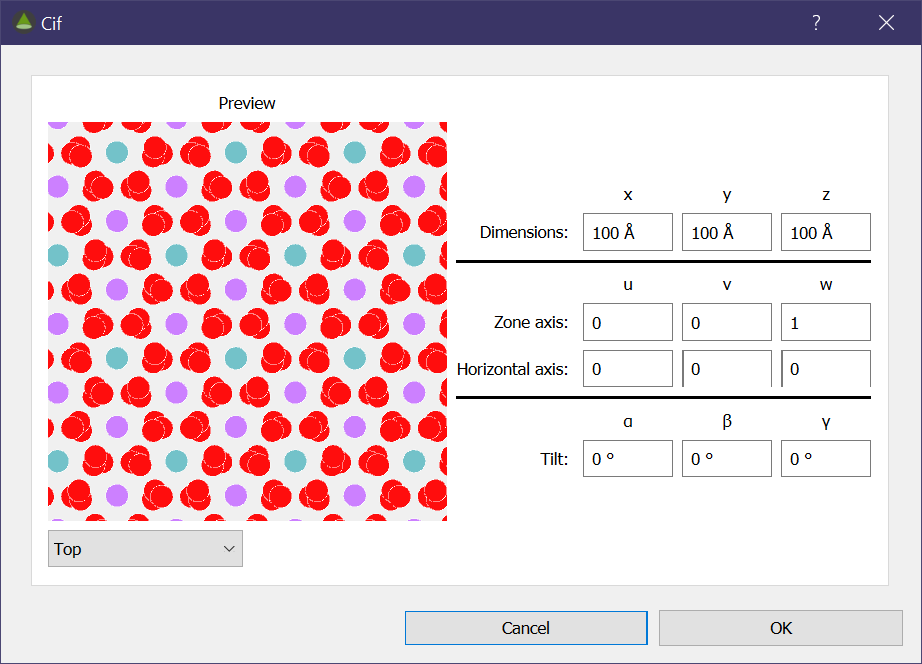
Figure Dialog used to configure OpenCL.
Once a valid .cif has been opened, the following dialog will be shown to construct the supercell. There is a live preview of the structure that will update as you change the inputs; the selection box underneath can be used to select the view direction (‘Top’ being the view of the incident electron beam). Here there are several options you can set.
- The z, y, z dimensions of the supercell
- The zone axis of the supercell
- The x-axis of the supercell. This will ignore any component along the zone axis.
- Tilts of the crustal structure applied about the simulation’s cartesian axes. These tilts are intended to be used for small perturbations.
Once you click OK, then the structure is loaded.
.xyz
The .xyz format is useful for defining non-periodic structures such as amorphous films, defects and interfaces. The simplicity of the format also means that generating files is relatively simple.
Basic .xyz files
At it’s simplest, the .xyz file has 3 main sections:
- the number of atoms in the structure
- comment section
- the atoms and their coordinates
The first line contains the number of atoms and the second line is reserved for comments. Every line after that represents an atom, in the form: A x y z, where A is the element symbol and x, y, z are the cartesian coordinates in Å. If you want to use the coordinates in nm, then include the term nm anywhere in the comments line (with whitespace either side). The use of nm then applied to all relevant units in the file (such as thermal vibrations).
Extra information
To add any more information, clTEM will look at the comments section as a sort of header section for the columns. This has defaults and multiple options to try and easily support as much as possible. They keywords that are looked for are A, x, y, z, occ, u, u1, u2 and u3. Any other terms will be ignored, so comments can be added (just make sure they don’t contain any of these terms separated by spaces).
A, x, y, z do not need to be stated, if none of them are found, it is assumed that they form columns 1-4 in the order shown. Other terms are then treated as columns 5 onwards, in the order they are written in the comments section.
Occupancy
Many structures require occupancies for the same sites. If the column occ is defined, clTEM will detect atoms that share the same site if they are placed on consecutive lines in the .xyz.
Thermal vibrations
Thermal vibrations are defined by the headers u, u1, u2 and u3 that define the isotropic vibration and then the vibration specific to each cartesian axis. u is set first, then any of u1, u2 or u3 will override the relevant component of u. u is defined here as the mean square displacement of the atom, <u²>, and will have the units of Ų (or nm² if the nm tag has been set).
Example file
Below is a small example file with occupancy and thermal parameters
6
occ u
Pb 0.0 0.0 0.0 1.0 0.0056
Pb 4.0 0.0 0.0 1.0 0.0056
Pb 4.0 4.0 0.0 1.0 0.0056
Pb 0.0 4.0 0.0 1.0 0.0056
Ti 2.0 2.0 0.0 0.8 0.0103
Zr 2.0 2.0 0.0 0.2 0.0048